



Inborn errors of metabolism can present with a variety of nonspecific signs and symptoms. While individually rare, as a class of disorders they are not uncommon and should be recognized and treated appropriately by emergency physicians.
Inborn errors of metabolism (IEMs) can present with a variety of nonspecific signs and symptoms. Acute metabolic decompensations can occur either in the perinatal period or later in childhood and are usually triggered by stressors such as fever, vomiting or prolonged fasting. A workup for metabolic disorders must be initiated alongside the standard diagnostics in the event of an unexpected deterioration in a full term baby after an uneventful perinatal period. IEMs result from enzyme deficiencies in important metabolic pathways. They are usually single gene disorders inherited in Mendelian patterns with some exceptions (eg, mitochondrial disorders). Symptoms arise due to accumulation of toxic substances (such as ammonia), failure of energy production systems, or accumulation of complex molecules in organelles.
Clinical Presentation
Newborns may be completely asymptomatic at birth, and it may take anywhere from hours to weeks for features to develop. The birthing process can itself be a trigger for the decompensation or it could be following the introduction of carbohydrates, proteins and lipids via feeds. Since they can be unresponsive to conventional therapies, obtaining a detailed history is extremely vital.
Vomiting can be a common presenting symptom. Children can become dehydrated from significant vomiting and it is more important to ascertain whether the child is able to maintain adequate intake of required calories. Neurological manifestations can range from decreased activity, altered mental status, lethargy and seizures to coma and death. Decreased activity can lead to poor feeding and therefore inadequate caloric intake, perpetuating a vicious cycle. Certain conditions may present with hypotonia or apneic episodes. Metabolic disorders are among the many possible etiologies for children presenting with seizures. Dysfunction of certain organ systems may also indicate an underlying metabolic condition. Jaundice, respiratory distress, failure to thrive, hiccups, and hypothermia should all raise suspicion for IEMs. Abnormal odor of the urine could be a possible manifestation of metabolic disorders.
As with most genetic disorders, eliciting a family history is important (eg, other affected family member, siblings, and multiple unexplained early deaths). Since most metabolic disorders are inherited in an autosomal recessive manner, a history of consanguinity would raise suspicion. Some disorders like OTC deficiency are X linked, and are less likely to cause a severe presentation in girls. The differential diagnoses for a child presenting with these symptoms include sepsis, cardiac dysfunction, encephalitis, altered mental status secondary to traumatic brain injury, ingestion, and electrolyte disturbances among others.
Physical Exam
A thorough physical exam may reveal the presence of syndromic features/dysmorphologic features that could narrow the diagnosis. For example, the presence macrocephaly, hypospadias, and rocker bottom feet could point the diagnosis in the direction of glutaric aciduria type 2.The exam should include evaluation of facial features, a detailed ophthalmologic exam, hearing evaluation, cardiac exam, abdominal exam for hepatosplenomegaly, neurological exam, and a genital exam. Possible abnormal features that may be observed include cataracts, cardiac murmurs, features of myopathy, hepatomegaly, and ascites. Rapid breathing could be an indication of efforts of compensate of metabolic acidosis as a consequence of urea cycle defects or hyperammonemia.
Investigations
The advancement of newborn screening has dramatically reduced the mortality and morbidity associated with many IEMs. It involves universal screening of all newborns through dried blood spots obtained through a heel prick at birth. The goal is to screen for treatable metabolic as well as some non-metabolic diseases at pre-symptomatic stages.
A battery of tests should be ordered when suspecting a metabolic condition including but not limited to - a blood gas, glucose, lactate, ammonia, LFTs, plasma amino acids, urine organic acids, plasma carnitine, plasma acylcarnitine, electrolytes, blood urea nitrogen, uric acid, complete blood count, and urine ketones (think rainbow tube set). Ammonia samples should always be drawn under the following parameters: free flowing sample, directly placed on ice and analyzed within 1 hour of drawing the sample. Plasma amino acids and urine organic acids will usually result much later, but should be drawn at presentation, as they are more likely to be abnormal and therefore diagnostic during an acute metabolic crisis. Consider obtaining CSF amino acids and CSF neurotransmitters if performing a lumbar puncture in the setting of neurological features.
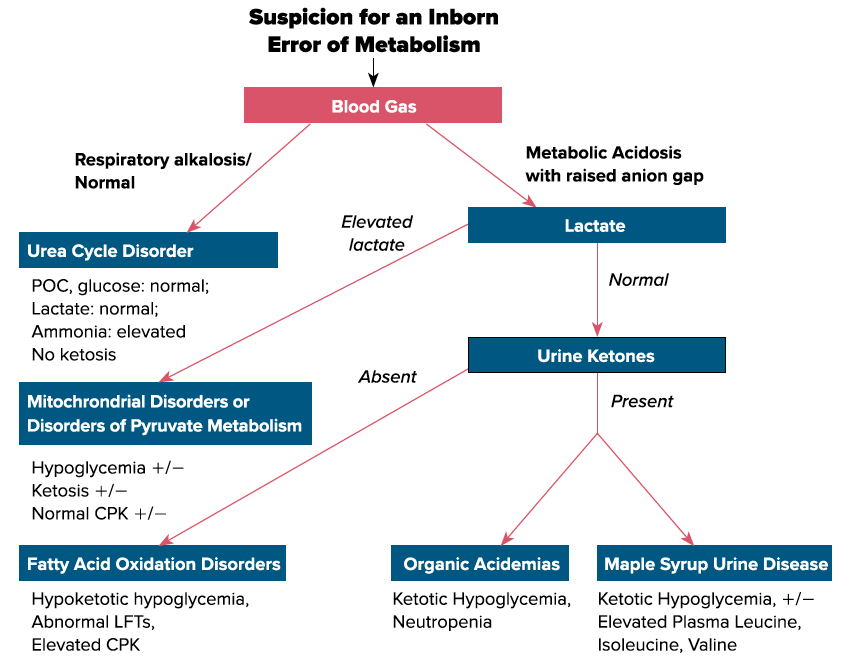
Management
In the presence of a known IEM, many families may carry a management protocol which is to be followed during acute illnesses. Dextrose containing fluids should be started immediately in most categories of IEMs to supplement the body’s metabolic needs, and blood tests to evaluate the metabolic status should be ordered. The primary metabolic provider should be consulted for further management sooner than later. As confirmation of a diagnosis can take time, the initiation of empiric treatment can be lifesaving. Intravenous dextrose should be initiated immediately and a 10% dextrose solution is the preferred via a peripheral IV. The IV fluids should be run at a rate enough to provide 120% of baseline caloric requirement. Age appropriate electrolytes should be included in the IV bag. Intralipids may also be used for additional calories; however this may be contraindicated in certain IEMs such as fatty acid oxidation disorders. It is important to note that the infusion of dextrose and lipids is for the provision of adequate caloric intake to prevent a catabolic state.
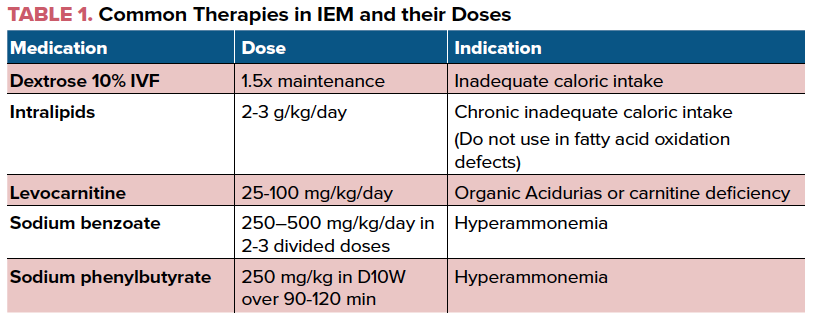
As the pathology of many metabolic disorders results from the accumulation of toxic metabolites in catabolic pathways, the initial aim of treatment is to prevent the breakdown of the substrates. Therefore, dietary intake of the implicated substrates (such as protein in urea cycle disorders or maple syrup urine disease) should be temporarily eliminated (NPO, special formula protein free or lactose free, ketogenic diet etc). Correct dehydration, metabolic acidosis, or electrolyte abnormalities. Consider sodium bicarbonate or acetate in the presence of significant acidosis. Eg: Hyponatremia should be promptly corrected in a maple syrup urine disease patient who could have cerebral edema secondary to leucine encephalopathy. Continue monitoring response to therapy and interventions.
The next step is to increase the excretion of the already accumulated toxic metabolites. Sodium benzoate and sodium phenylbutyrate or Ammunul may be used to decrease plasma ammonia levels. The presence of refractory metabolic derangements such as hyperammonemia or lactic acidosis may be indications for hemodialysis as the renal excretion rate may not be fast enough to prevent secondary end organ damage. Levocarnitine is widely used in primary carnitine uptake deficiency, organic acidemias, fatty acid oxidation disorders and other metabolic disorders. This medication helps to transport fatty acids into the mitochondria and also aids in elimination of organic acids by forming carnitine esters.
Conclusion
IEMs are individually rare, but as class of disorders they are not uncommon. Each IEM is a unique clinical entity with a characteristic biochemical defect. However, they share a permutation of metabolic aberrations, and based on these certain generic management strategies can be formed. As metabolic doctors are usually only available at large academic centers, emergency physicians are often at the forefront to identify and treat these children. Maintaining a high index of suspicion, ordering the initial gamut of biochemical testing in a timely manner and being well versed with basic treatment strategies goes a long way in reducing the mortality and morbidity associated with these diseases.
This article aims to discuss cardinal strategies in the acute management of IEMs. However, physicians can also refer to the following resources (GeneReviews, New England Consortium of Metabolic Programs) for further detailed management.
References
1. Bennett E, Hummel K, Smith A, Longo N. Acute Presentation and Management of the Encephalopathic Child With an Undiagnosed Inborn Error of Metabolism. J Emerg Med. 2019;56(1):e5-e8.
2. El-Hattab AW. Inborn Errors of Metabolism. Clin Perinatol. 2015;42(2):413-439.
3. Romão A, Simon PEA, Góes JEC, et al. Initial Clinical Presentation In Cases Of Inborn Errors Of Metabolism In A Reference Children's Hospital: Still A Diagnostic Challenge. Rev Paul Pediatr. 2017;35(3):258-264.